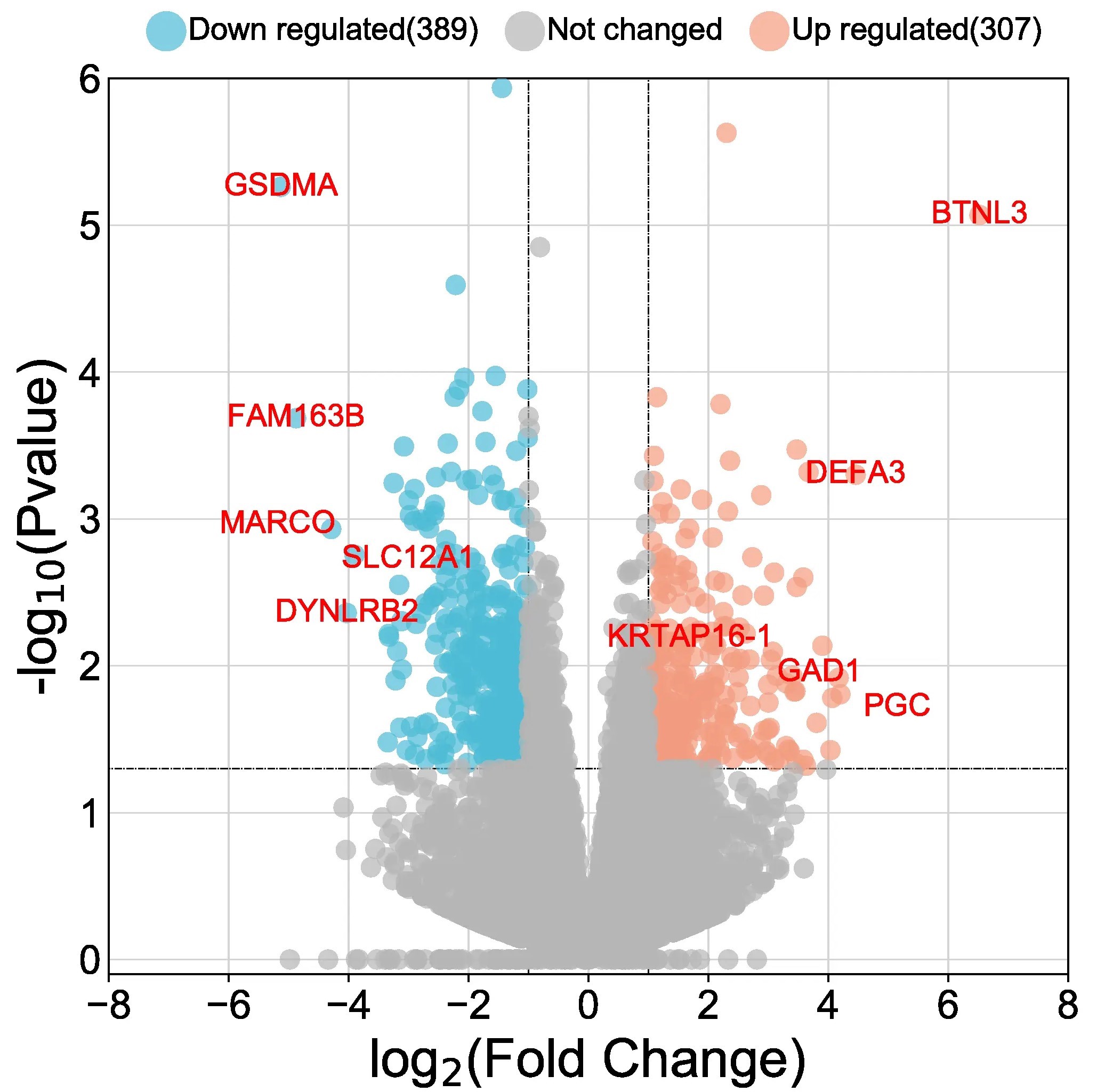
DESeq2
A basic task in the analysis of count data from RNA-seq is the detection of differentially expressed genes. The count data are presented as a
table which reports, for each sample, the number of sequence fragments that have been assigned to each gene. Analogous data also arise for
other assay types, including comparative ChIP-Seq, HiC, shRNA screening, and mass spectrometry.
An important analysis question is the quantification and statistical inference of systematic changes between conditions, as compared to
within-condition variability. The package DESeq2 provides methods to test for differential expression by use of negative binomial generalized
linear models; the estimates of dispersion and logarithmic fold changes incorporate data-driven prior distributions.
Love MI, Huber W, Anders S (2014). “Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2.” Genome Biology, 15,
550. doi:10.1186/s13059-014-0550-8.